DOGMA (DOmain-based General Measure for transcriptome and proteome quality Assessment) is
a program to estimate the quality of a given set of sequences. A typical use case is to assess the quality of a newly assembled transcriptome or the newly annotated proteome of
a genome. On this page you will find information concerning the usage of the DOGMA web server.
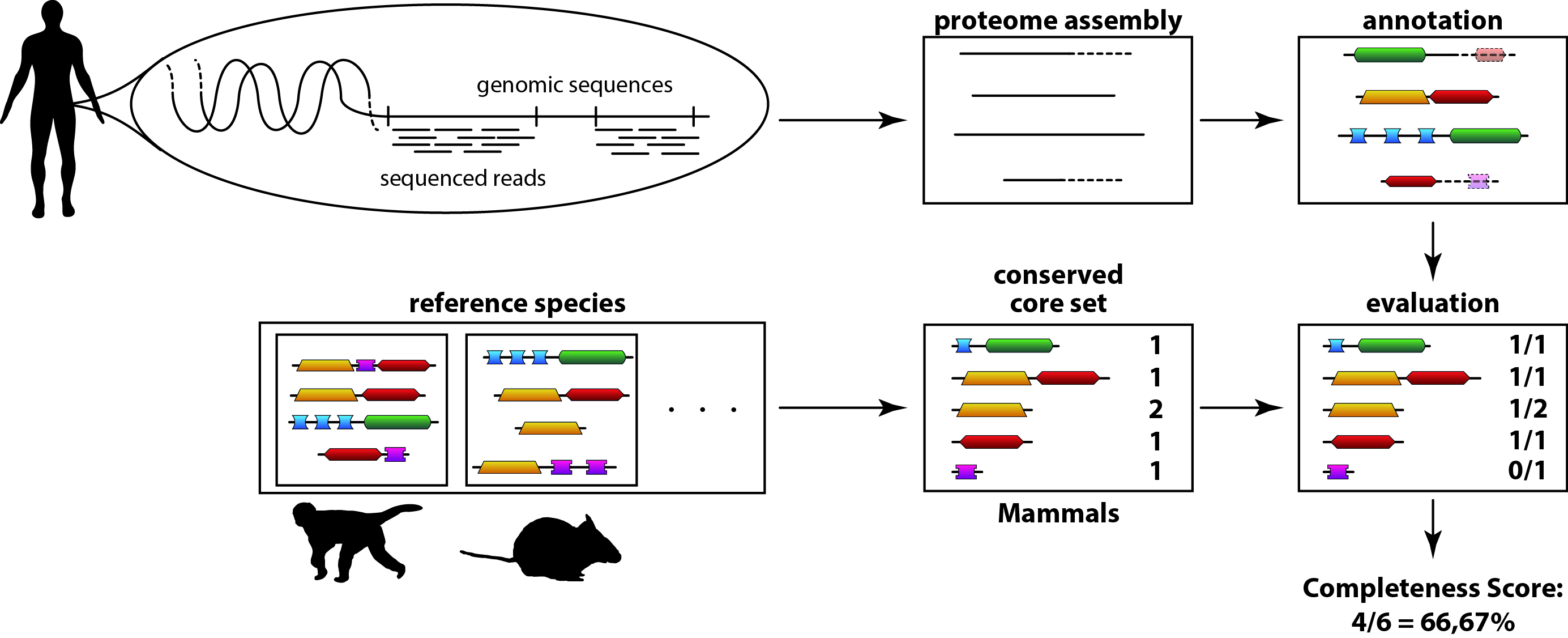
The principle of the DOGMA algorithm is simple: A known set of conserved domain arrangements (CDAs) are compared with the
annotated domain arrangements of a proteome/transcriptome to analyse. DOGMA returns the percentage of found CDAs,
indicating the quality of the analysed proteome/trancriptome.
Overview of the DOGMA quality assessment algorithm
For more information on how the algorithm works, please have a look at the
publication of DOGMA
The web service
This chapter describes the web server functions
Starting DOGMA
Input
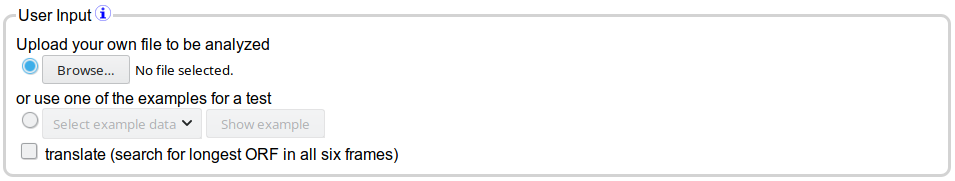
DOGMA provides a simple interface to determine the data to analyse:
The DOGMA input section
The easiest and fastest is to directly upload your annotation file. The highest accuracy and most detailed results can be expected when using
Pfam annotations of your isoform free proteome. In case the proteome has isoforms it is recommended to create a new file containing only the longest ones.
We additionally accept RADIANT annotations as well, but please
be aware of the current limitations when using RADIANT (see RADIANT for more information).
If you provide a sequence file in FASTA format containing proteins or spliced genes instead, the server will run RADIANT first to annotate the sequences with
domains. RADIANT is a very fast domain annotation program that in general works very well together with DOGMA. However,
when annotating species whose domains are very distinct from the ones in the Pfam database, RADIANT might perform poorly
and therefore lower the DOGMA score (see RADIANT for more information). It is not possible to analyse whole genomes/assemblies as only a single ORF per sequence will be predicted.
You have as well the option to use one of the provided example files for a test run. After having selected an example file, the "Show example" button can be used to
open the file in a text editor of your choice. If you press "Run" DOGMA will be executed with the selected example file. You can simply use the default parameters for these files.
If you provide nucleotide sequences you have to check the translate button. It will translate the sequences into amino acids by predicting
the longest ORF in all 6 frames.
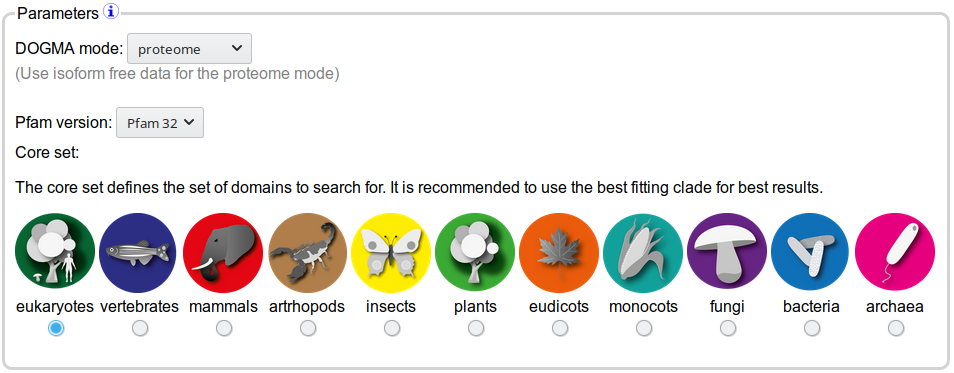
Parameters
This section will explain the different parameters that you can choose to influence the behaviour of DOGMA.
The DOGMA parameter section
DOGMA mode
The DOGMA mode influences how the DOGMA score is calculated. The proteome mode provides better accuracy estimations
because it can take into account the number of found CDAs for each CDA type. Therefore, an isoform free sequence set is needed for a
correct calculation. As there is no
general way to identify them you will have to provide one. The transcriptome mode on the other hand completely ignores the
exact number of specific CDAs and only checks for presence or absence.
Pfam version
If you provide the domain annotation directly, you should choose here the same Pfam version you used to annotate your
sequences. If you provide sequences, this choice will determine which Pfam version will be used to annotate them.
Choice of core set
The core set defines the set of domains that is compared to the sequences to be analysed. We provide precomputed core
sets for different clades. For best results choose the most specific core set available as it will contain a higher number of
domains to which can be compared and therefore provides a higher accuracy.
Result interpretation
The result page of DOGMA will display the DOGMA analysis results. Additionally, the used parameters are shown.
DOGMA score
The main result of DOGMA is summarized in a table. It compares the number of found conserved domain arrangements (CDAs)
with the expected number from the core set. Separate values for CDAs of length 1,2 and 3 are provided. Furthermore, the last
row shows the total percentage, the DOGMA score.
CDAsize
#Found
#Expected
%Completeness
1
1036
1051
98.57
2
428
441
97.05
3
161
167
96.41
Total
1625
1659
97.95
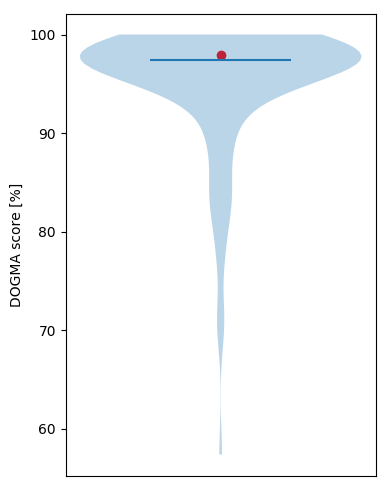
Additionally, a graphic relates your DOGMA score to a larger set of known proteomes. The graphic compares the quality of
your proteome/transciptome (red dot) to a precomputed set of DOGMA scores which belong to the same
clade as the chosen core set. With the exception of archaea, the proteomes for the comparisons were all taken from ensembl
databases. The proteomes for the archaea comparison are from different sources.
An example of the comparison result. The red dot shows how your data compares to
a set of known proteomes of the same core set. The vertical blue line denotes the median. The width of the blue area
denote the density of proteomes with this DOGMA score.
Partial domain analysis
This analysis is only available when a PFAM annotation was provided. The partial domain analysis calculates the general occurrence
of partial domains in your proteome set and is not limited to the domains in the core set. A domain is considered to be partial
if its length is shorter than half of the expected size.
An example can be seen below:
fraction of partial domains:
3.99%
number of partial domains:
1750
total number of domains:
43871
coverage threshold:
0.5
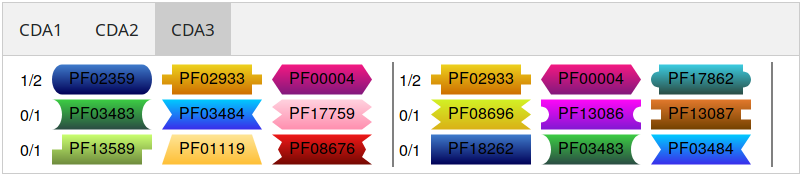
Missing CDAs
This section displays a detailed overview of the missing CDAs of your data. They are sorted by size. If you hover over a
domain it will show you the name of the Domain. The domain itself is a link to the Pfam entry of it.
Listing of missing CDAs of size 3.
RADIANT
RADIANT is a program we are currently developing. It allows to annotate a proteome in a few minutes with domains. The
very fast annotation allows its usage on a web server. However, while in the majority of the cases the domain annotation
is actually very good and has very little influence on the DOGMA score, in some cases it does perform poorly which influences (decreases)
the DOGMA score although the proteome/transcriptome is of good quality. We therefore recommend RADIANT to be used only
to get a first fast impression of the quality and perform afterwards a check with a Pfam annotation (especially in cases when
the DOGMA score is low).
How to cite
If you used DOGMA in your project please cite our publication:
Elias Dohmen, Lukas P.M. Kremer, Erich Bornberg-Bauer, and Carsten Kemena, DOGMA: domain-based transcriptome and proteome quality assessment, Bioinformatics (2016) 32 (17): 2577-2581. doi:10.1093/bioinformatics/btw231